RNA-protein complex refinement using AI modeling and docking
This article explains a workflow for refining protein-RNA complexes by combining AI-based structural models with flexible docking and enhanced sampling.
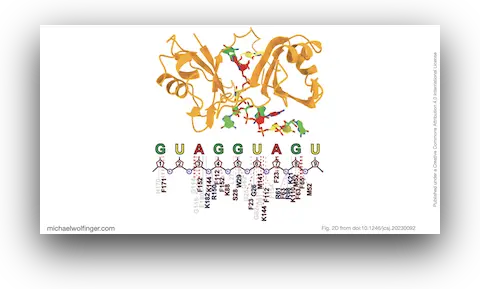
Modeling protein-RNA complexes remains difficult even when reasonably good structures for the individual components are available. The hardest part is often not generating a starting model, but refining the interface in a way that captures flexible RNA segments and produces a biologically plausible binding geometry.
This study addresses that problem with a two-step workflow. First, AlphaFold2 was used to generate a structural model for the RNA-binding domains of the human Musashi-1 (MSI1) protein. Second, the resulting model was refined in the presence of RNA using flexible docking based on parallel cascade selection molecular dynamics (PaCS-MD). The point of the method is not simply to place RNA near a protein surface, but to sample interface rearrangements that matter for complex formation.
Musashi-1 is a useful test case because its RNA recognition has been studied experimentally, which means the resulting models can be checked against known interaction patterns. In the refined complexes, the analysis recovered a core set of residues and nucleotides that are consistent with previous work on MSI1-RNA recognition. That matters more than raw structural novelty. A refinement method is only useful if it recovers contacts that make biochemical sense.
Compared with a more standard template-based workflow built around Phyre2, the PaCS-MD approach produced better-supported association complexes in this system. The main reason is that enhanced sampling gives flexible RNA regions more room to explore realistic conformations during docking, instead of forcing the final model to depend too heavily on manual assembly or rigid starting assumptions.
The method still has clear limits. It depends on having useful initial structural information, and the quality of the final complex remains tied to the quality of both the starting model and the sampling protocol. This is not a general solution to protein-RNA structure prediction from sequence alone. It is a refinement strategy that becomes valuable when there is already enough structural context to make flexible docking meaningful.
That makes the study best understood as a methods contribution. It shows that AI-derived protein models can be combined with enhanced-sampling docking to improve protein-RNA complex refinement, at least for systems like MSI1 where independent evidence exists for the binding interface. For researchers working on RNA-binding proteins, this is a more realistic and useful claim than broad promises about drug discovery.
I place that point in a wider modeling context in What AI can and cannot do for RNA structure and RNA-protein modeling.
If your lab or company needs an external review of an RNA-protein modeling workflow, a structure-guided design problem, or a docking strategy, I also offer focused advisory support through my services page. A Structural Refinement Technique for Protein-RNA Complexes Using a Combination of AI-based Modeling and Flexible Docking: A Study of Musashi-1 Protein Theoretical studies on RNA recognition by Musashi 1 RNA–binding protein Musashi Binding Elements in Zika and Related Flavivirus 3’UTRs: A Comparative Study in SilicoCitation
Nitchakan Darai, Kowit Hengphasatporn, Peter Wolschann, Michael T. Wolfinger, Yasuteru Shigeta, Thanyada Rungrotmongkol, Ryuhei Harada
B. Chem. Soc. Jpn. (2023)See Also
Nitchakan Darai, Panupong Mahalapbutr, Peter Wolschann, Vannajan Sanghiran Lee, Michael T. Wolfinger, Thanyada Rungrotmongkol
Sci. Rep. 12:12137 (2022) | doi:10.1038/s41598-022-16252-w | PDF | Figures
Adriano de Bernardi Schneider, Michael T. Wolfinger
Sci. Rep. 9(1):6911 (2019) | doi:10.1038/s41598-019-43390-5 | PDF | Figures