Automated lineage designation from viral genomic data
This study describes an automated framework for lineage designation from phylogenetic and genomic data, designed to scale to very large viral datasets while remaining consistent and interpretable.
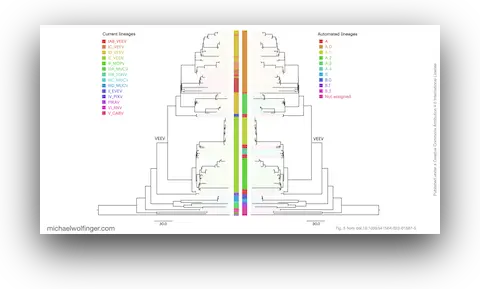
Lineage designation is partly a biological question and partly an information-management problem. Once viral sequencing reaches very large scale, as it did for SARS-CoV-2, manual or community-curated naming systems become increasingly difficult to maintain. Delays accumulate, criteria become harder to apply consistently, and the workload grows faster than the nomenclature process can keep up.
This study presents Autolin, a heuristic framework for automated lineage designation from phylogenetic and genomic data. The main contribution is not a claim that expert curation should disappear, but that a simple and explicit rule-based system can produce lineage assignments at a scale that is difficult to sustain manually.
That matters because lineage systems are only useful when they are both interpretable and sustainable. An automated framework can evaluate very large trees, apply the same criteria repeatedly, and generate designations without depending on ad hoc proposal cycles. In practice, that means faster turnaround and more consistent behavior across datasets with millions of sequences.
Another useful feature of the framework is that it allows prioritization of particular mutations or genes. That makes the method flexible enough to reflect biological or epidemiological priorities rather than treating all sequence variation as equally informative. In other words, the system is not just scalable. It can also be tuned to highlight the parts of a genome that matter most for a given pathogen or surveillance context.
The paper is careful about scope. Automated lineage designation is not the same thing as biological interpretation, and no heuristic can remove the need for expert judgment altogether. What it can do is provide a consistent baseline classification system that remains usable as genomic datasets continue to grow. The fact that the method produces lineage partitions similar to existing curated systems across multiple viruses makes that claim much more credible.
For genomic epidemiology, this is the real value of the framework. It gives researchers a practical way to maintain phylogeny-based nomenclature under conditions where purely manual designation becomes increasingly fragile. That is useful not only for SARS-CoV-2, but also for other rapidly sampled viral systems where scale has already become the defining constraint.
This is also one of the clearest cases in which automation addresses a real bottleneck without pretending to resolve molecular mechanism by itself. A framework for automated scalable designation of viral pathogen lineages from genomic dataCitation
Jakob McBroome, Adriano de Bernardi Schneider, Cornelius Roemer, Michael T. Wolfinger, Angie S. Hinrichs, Aine N. O’Toole, Chris Ruis, Yatish Turakhia, Andrew Rambaut, and Russell Corbett-Detig
Nature Microbiol. 9:550–560 (2024) | doi:10.1038/s41564-023-01587-5 | PDF