When sequence conservation is not enough to find functional RNA structure
Functional RNA structures are often preserved long after primary sequence similarity becomes weak or misleading. In those cases, comparative analysis has to look for conserved architecture, compensatory change, and shared structural logic rather than sequence identity alone.
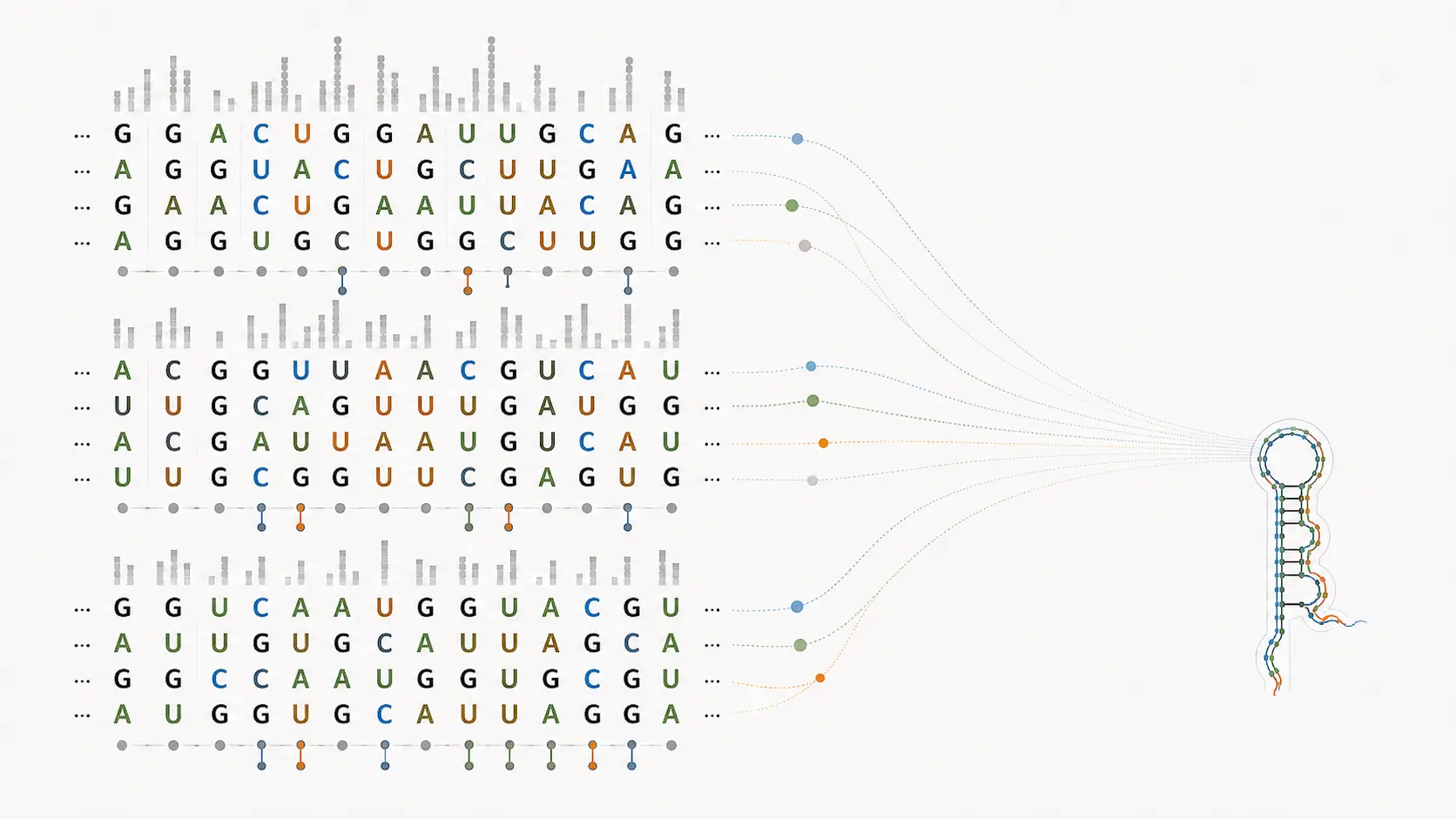
Sequence conservation is a useful starting point in comparative biology, but it can be a poor guide to functional RNA structure. Structured RNAs may remain under selection at the level of base pairing, topology, or higher-order organization even while their primary sequence drifts. A sequence-only view will miss part of that signal.
The point becomes especially clear in viral untranslated regions. In many RNA viruses, the non-coding parts of the genome do not merely separate coding segments or stabilize transcript ends. They carry structured elements that influence replication, cyclization, nuclease resistance, host adaptation, and regulatory timing. Those functions can remain conserved even when the underlying sequence has changed substantially.
Comparative RNA analysis therefore has to ask a different question. Instead of following sequence identity alone, it has to examine whether the same architecture has been preserved. Compensatory substitutions, recurring motif arrangements, and conserved mechanistic roles become more informative than local sequence similarity by itself.
The flavivirus 3'UTR provides a particularly good example. In Comparative genomics of flavivirus 3' UTR RNA structures, the point is not simply that these viruses contain structured RNA. The deeper result is that lineages with very different sequence histories can still preserve exoribonuclease-resistant RNAs and other functional modules through shared structural organization. What is conserved is often the mechanism, not the exact wording of the sequence.
The same logic becomes even clearer in the mosquito-borne branch. Functional RNA structures in the 3'UTR of Mosquito-Borne Flaviviruses shows that these genomes are built from a recurring structural vocabulary, including xrRNAs, dumbbells, and terminal stem-loops, yet the number, arrangement, and detailed realization of those elements vary across groups. A sequence-based comparison alone would flatten that picture. Structural comparison reveals the lineage-specific architecture.
Functional RNA elements are not always portable as simple motifs. An xrRNA is not defined by a short consensus sequence. It is defined by a fold that creates a physical barrier to exonuclease progression. The same is true more broadly for many structured elements in viral genomes, whose activity depends on geometry and context.
Tick-borne flaviviruses offer a useful intermediate case. In RNA structure conservation and molecular epidemiology of TBEV, the central observation is that the 3'UTR varies, but not arbitrarily. The variable region still draws from a limited structural vocabulary. That means lineage diversification is better understood as a remodeling of architecture than as unconstrained sequence drift.
The same principle also explains why newly discovered lineages can be so informative. In Mpulungu virus and unique xrRNAs in a novel African tick flavivirus and the later Xinyang flavivirus clade paper, the genomes look unusual at the sequence level, yet the untranslated regions still preserve recognizable structured-RNA strategies. That kind of result would be very hard to see if one were only searching for clean sequence conservation.
This is one of the more important conceptual shifts in RNA virology. It changes what counts as homology, what counts as divergence, and what kinds of questions can be asked about function. Sequence divergence does not imply that structure has been lost. In many cases, the function has been re-encoded in a different sequence realization.
When the biology depends on structured RNA, sequence alignment is usually the beginning of the analysis rather than the end. Interpretation becomes much stronger once covariation, consensus folding, experimental validation, and comparative architecture are considered together.