Bacterial transcription start site annotation from dRNA-seq data
TSSAR introduced statistically grounded, automated annotation of bacterial transcription start sites from dRNA-seq data and packaged it as both a RESTful web service and a standalone tool.
Identifying bacterial transcription start sites from differential RNA-seq data used to be a painfully manual task. Researchers would inspect mapped reads in genome browsers, compare TEX-treated and untreated libraries by eye, and then decide which positions looked like genuine primary transcript starts. That process was slow, difficult to reproduce, and inevitably shaped by user-specific thresholds and biases.
TSSAR was designed to solve exactly that problem. The aim was not just to call more TSS automatically, but to provide a statistically principled way to do so. Differential RNA-seq enriches primary transcripts by treating one library with terminator exonuclease, so the key signal is an excess of read starts at a genomic position in the TEX-treated sample relative to the untreated control. TSSAR turns that intuition into a formal model instead of leaving it at the level of browser-based pattern recognition.
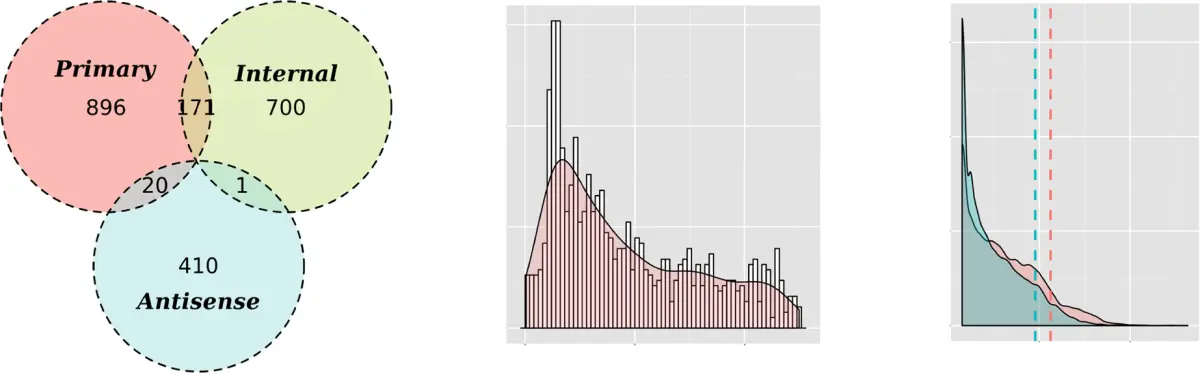
The methodological core is a local count model for read starts within transcriptionally active regions. Counts in individual libraries are modeled in a way that leads naturally to a Skellam-distributed difference between treated and untreated start counts. That provides a direct statistical basis for deciding whether an observed enrichment is likely to reflect a genuine primary transcript start rather than noise. The output is not just a list of coordinates, but an annotated classification into primary, internal, antisense, and orphan signals that can be used downstream.
That combination of statistics and annotation logic is what made the tool useful in practice. TSSAR does not simply replace manual curation with a fixed arbitrary cutoff. It automates the decision process while still respecting the structure of dRNA-seq experiments. In the paper, the method is benchmarked against manual annotations and simple cutoff-based alternatives, and it performs substantially better at recovering both curated Helicobacter pylori TSS annotations and experimentally validated sites.
One reason the paper still feels current is its software architecture. TSSAR was not designed as two separate products, one local and one web-based. Instead, the workflow was intentionally split across a local client and a RESTful web service that depend on each other. The local component handles the preprocessing of mapped NGS data, while the processed data are then submitted to the service for the actual TSS-oriented statistical analysis and annotation steps. In other words, the client and service form a coupled pipeline rather than two interchangeable access modes.
That distinction matters, because it reflects how the method was meant to be used in practice. The computationally and format-specific preprocessing stayed close to the user's data and local environment, while the web service centralized the analysis logic and reporting layer. This design reduced the burden of reproducing a fairly specialized workflow in many separate installations, but it also means the local and remote components should not be described as independently useful alternatives. The architecture is integrated by design.
That architectural decision may be just as important as the underlying statistics. A method only has impact if people can actually use it, and TSSAR addressed that by combining client-side preprocessing with a service-backed analysis workflow. That is one reason it remained relevant well beyond the immediate paper.
The broader biological significance is also straightforward. Better TSS annotation improves our view of bacterial transcriptome architecture: operon boundaries, antisense transcription, condition-specific initiation, and the regulatory logic of promoter usage. In that sense, TSSAR is not just a niche utility for one sequencing protocol. It is infrastructure for studying how bacterial gene regulation is organized at the transcript level.
This paper is clearly a different topic from RNA folding or landscape analysis, but the underlying engineering mindset is similar. The interesting part is not only the biology. It is also how to turn a noisy, high-dimensional data source into a usable and reproducible computational workflow. That is exactly the kind of problem where good statistical assumptions, careful software design, and sensible interfaces make the difference between a promising idea and a method that people keep using. TSSAR: TSS annotation regime for dRNA-seq dataCitation
Fabian Amman, Michael T. Wolfinger, Ronny Lorenz, Ivo L. Hofacker, Peter F. Stadler, Sven Findeiß
BMC Bioinformatics 15:89 (2014) | doi: 10.1186/1471-2105-15-89 | PDF