Latest article
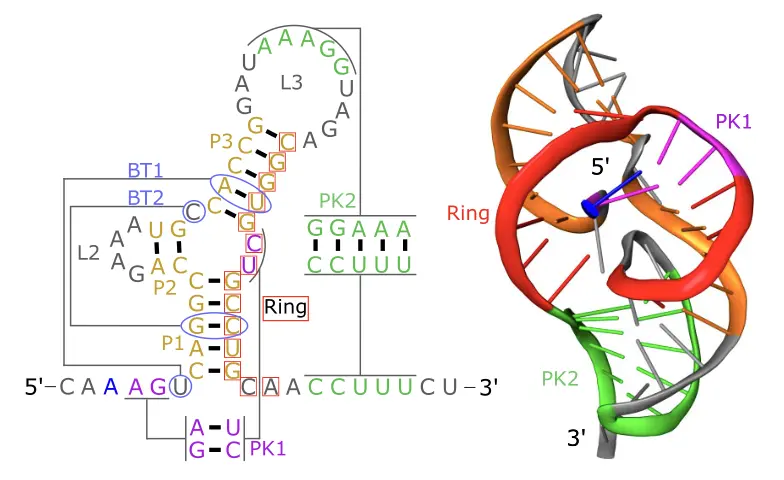
Rational design of mechanically active RNAs
This Nucleic Acids Research paper shows that synthetic xrRNAs can be designed from topological rules and validated experimentally as mechanically active RNAs.
Earlier posts
- What AI can and cannot do for RNA structure and RNA-protein modeling
- Why kinetic folding matters in RNA design
- From structure to function in Musashi-RNA complexes
- Bayesian approximation of RNA folding times
- KinPFN for RNA folding kinetics
- 3' UTR-biased siRNA production in an insect-specific flavivirus
- Xinyang flavivirus and a likely tick-only orthoflavivirus clade
- Automated lineage designation from viral genomic data
- A conserved G-quadruplex in the Zika virus 3' terminal region
- RNA-protein complex refinement using AI modeling and docking
page 1 | older articles »