Caveats in deep learning for RNA secondary structure prediction
This paper shows that many deep learning models for RNA secondary structure prediction learn dataset bias more readily than RNA folding rules, and explains why that matters for the future of AI in RNA biology.
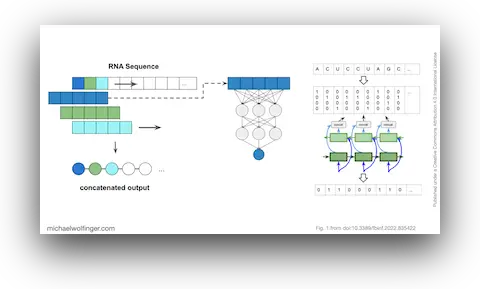
Deep learning for RNA secondary structure prediction has an obvious appeal. If neural networks can infer structure directly from sequence, perhaps they can move past some of the limitations of classical thermodynamic folding. That promise has made the area popular, but it has also brought in a familiar problem from other parts of machine learning. Strong benchmark numbers can hide the fact that a model has learned properties of the dataset rather than properties of the underlying biology.
That is the central concern of this paper. Instead of asking only whether a model performs well on a standard test split, we ask what it has actually learned. Has it captured transferable principles of RNA folding, or has it mostly memorized the structural biases of the RNAs it saw during training? For RNA structure prediction, that distinction matters a great deal. The real task is not to recognize another tRNA-like example from a familiar family. It is to say something useful about RNAs with new sequence-structure relationships.
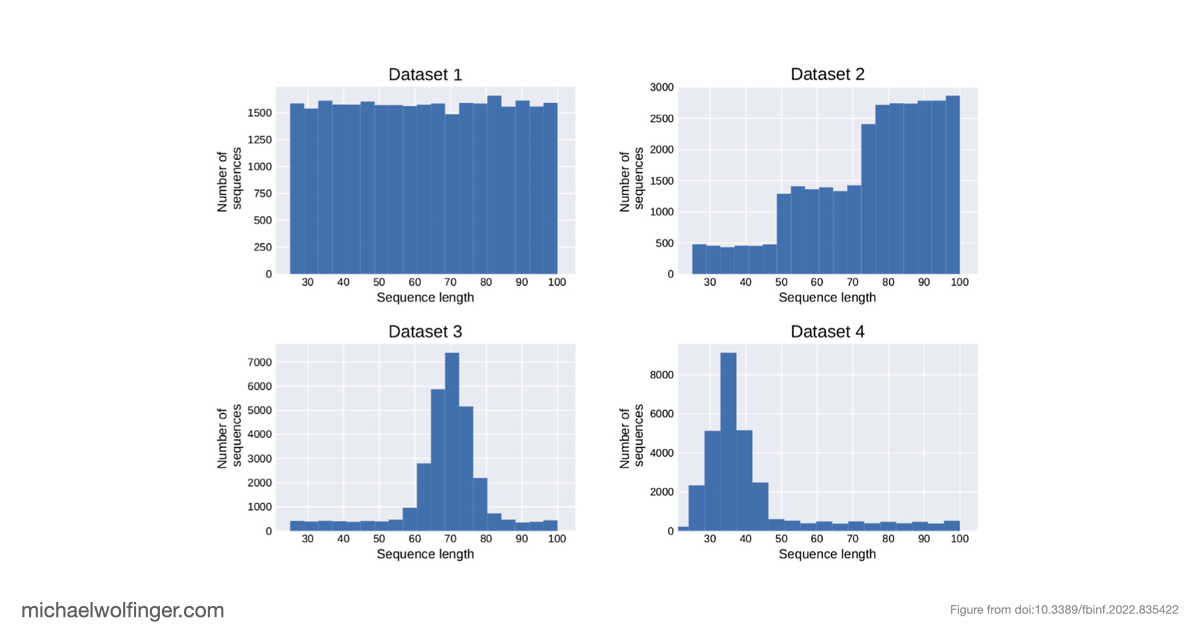
The methodological approach is deliberately controlled. We use inverse RNA folding to generate synthetic datasets with known structural properties, which lets us vary the amount of bias in the training data instead of merely inheriting whatever bias happened to be present in a benchmark collection. That setup makes it possible to compare model behavior on "more of the same" against genuinely novel structural patterns. The paper is not simply another machine-learning benchmark. It is a stress test for generalization.
The result is sobering but informative. When neural networks are trained on biased datasets, they can perform surprisingly well on held-out sequences that fold into familiar classes of structures. Once the test set contains structures outside those familiar patterns, performance drops sharply. The models generalize across sequence variation far more readily than they generalize across structural novelty. That is a warning sign for anyone hoping to use deep learning as a drop-in replacement for biophysical RNA folding models.
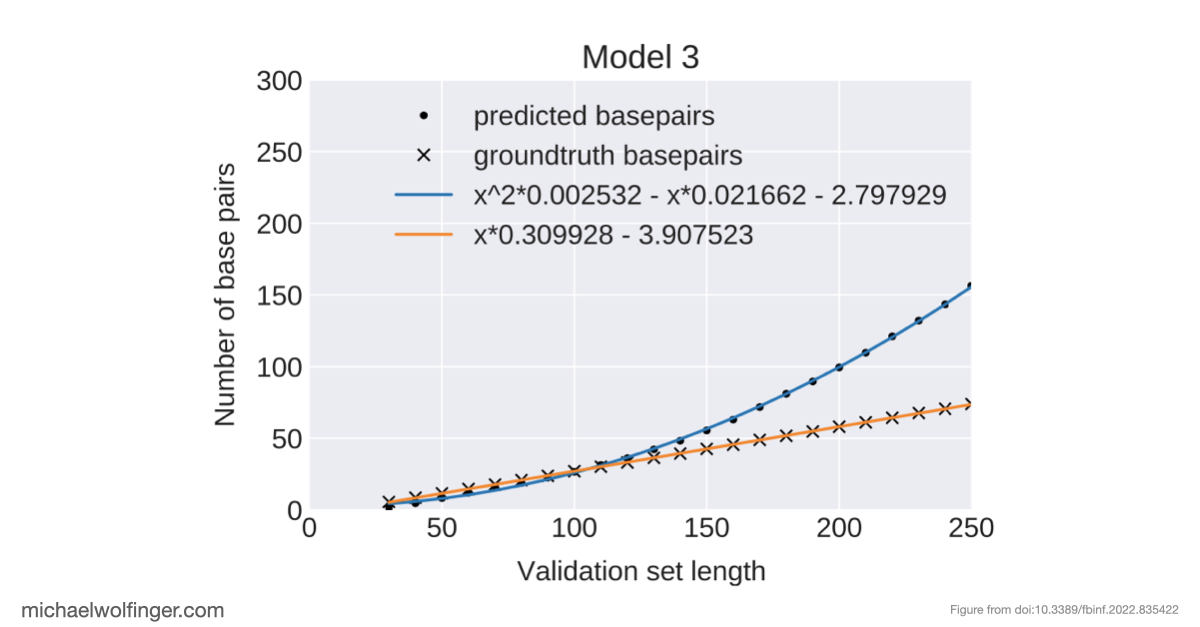
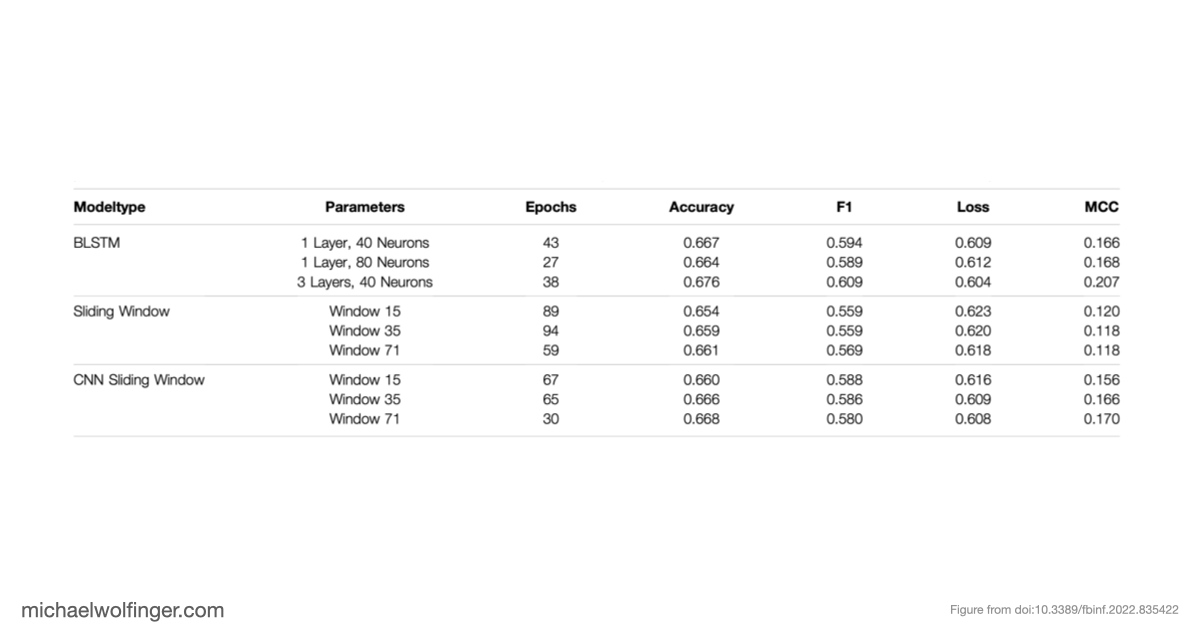
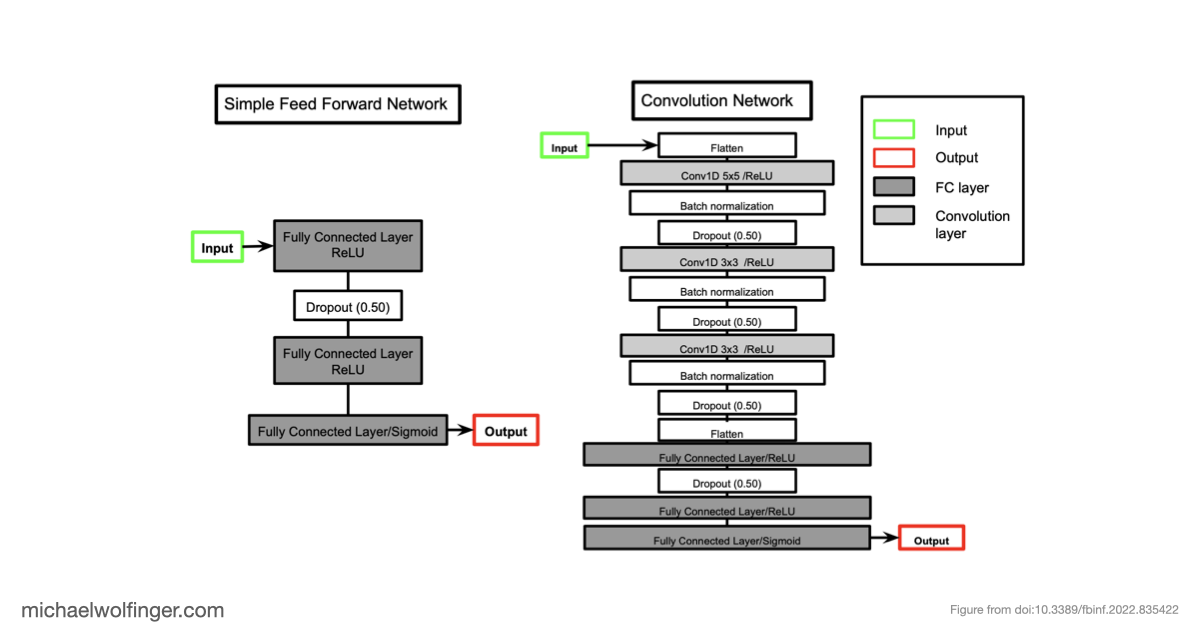
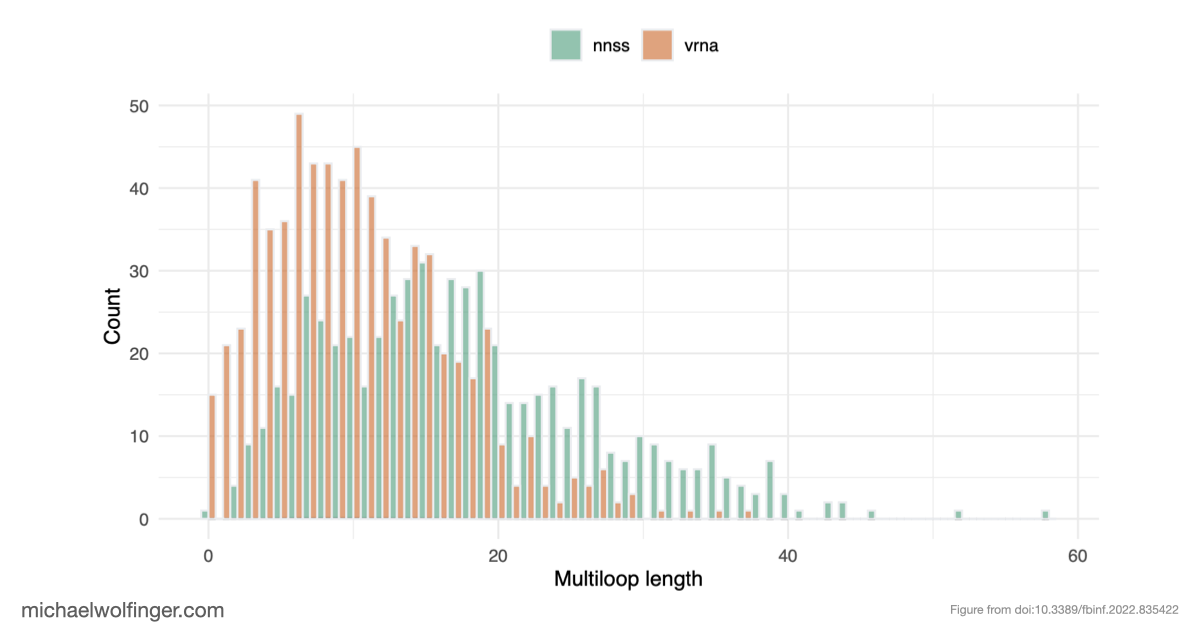
An equally important observation is that removing dataset bias does not magically solve the problem. Even on unbiased synthetic data, several architectures struggle to recover basic structural constraints reliably. Some models predict pairing patterns whose scaling with sequence length is inconsistent with valid secondary structures. Others produce artifacts that resemble pseudoknots or base triples even when the ViennaRNA-style ground truth does not contain such features at all. These are not minor numerical errors. They point to a mismatch between model output and the combinatorial rules that define the object being predicted.
I still find the article important years later because it is not an anti-AI paper. It is a paper about technical honesty. If machine learning is going to contribute meaningfully to RNA biology, evaluation has to distinguish memorization from mechanism. The models also have to respect the structural constraints of RNA rather than merely fitting correlations in a benchmark. The work argues, implicitly, for approaches that combine learning with stronger priors, explicit structure constraints, or experimental information instead of assuming that larger networks alone will fix the problem.
The related practical question is when a structure predictor explains a mechanism well enough to influence an experimental decision. The answer depends less on whether a model is branded as AI and more on whether the evidence is robust, interpretable, and proportionate to the cost of the next step.
For a broader assessment of how machine learning enters RNA biology beyond secondary structure prediction, see What AI can and cannot do for RNA structure and RNA-protein modeling.
Readers who arrive here from an AI angle may also want to look at some of my other work from the opposite direction. In Predicting RNA structures from sequence and probing data, I discuss how experimental structure probing can be integrated with computational prediction. In Conserved RNA regulatory switches in living cells, the focus shifts to transcriptome-scale structural ensembles and experimentally anchored regulatory switches. If you are more interested in dynamic folding than static structure, co-transcriptional RNA-ligand interaction dynamics shows the kind of mechanistic modeling that remains hard to replace with black-box prediction alone. Caveats to deep learning approaches to RNA secondary structure predictionFigures and Data


Citation
Christoph Flamm, Julia Wielach, Michael T. Wolfinger, Stefan Badelt, Ronny Lorenz, Ivo L. Hofacker
Front. Bioinform. 2:835422 (2022) | doi:10.3389/fbinf.2022.835422 | PDF