RNA–RNA interaction analysis of Japanese encephalitis virus
This study combines computational and biophysical analysis to characterize a long-range RNA-RNA interaction in Japanese encephalitis virus and to test the role of the conserved cyclization sequence.
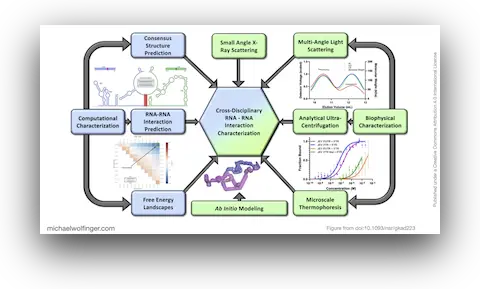
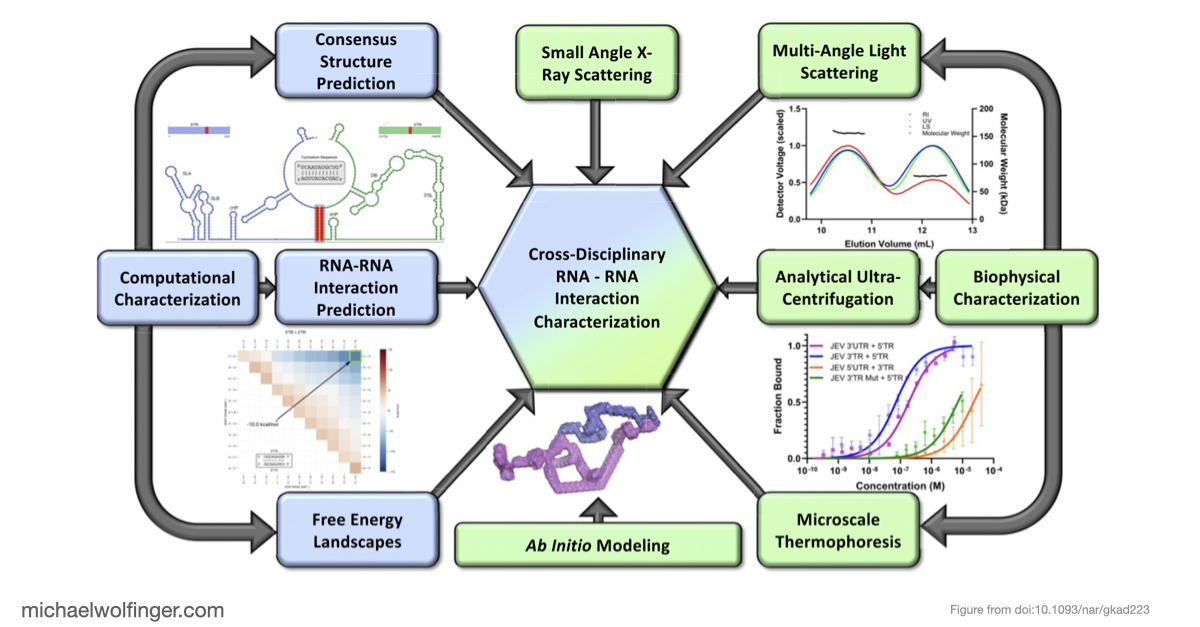
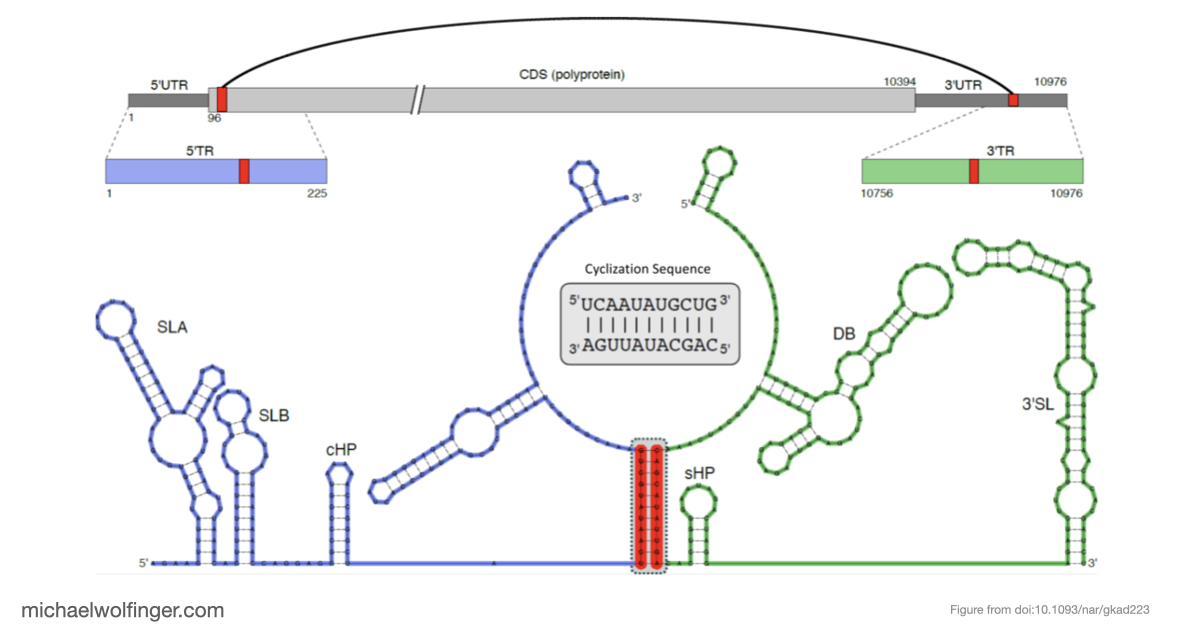
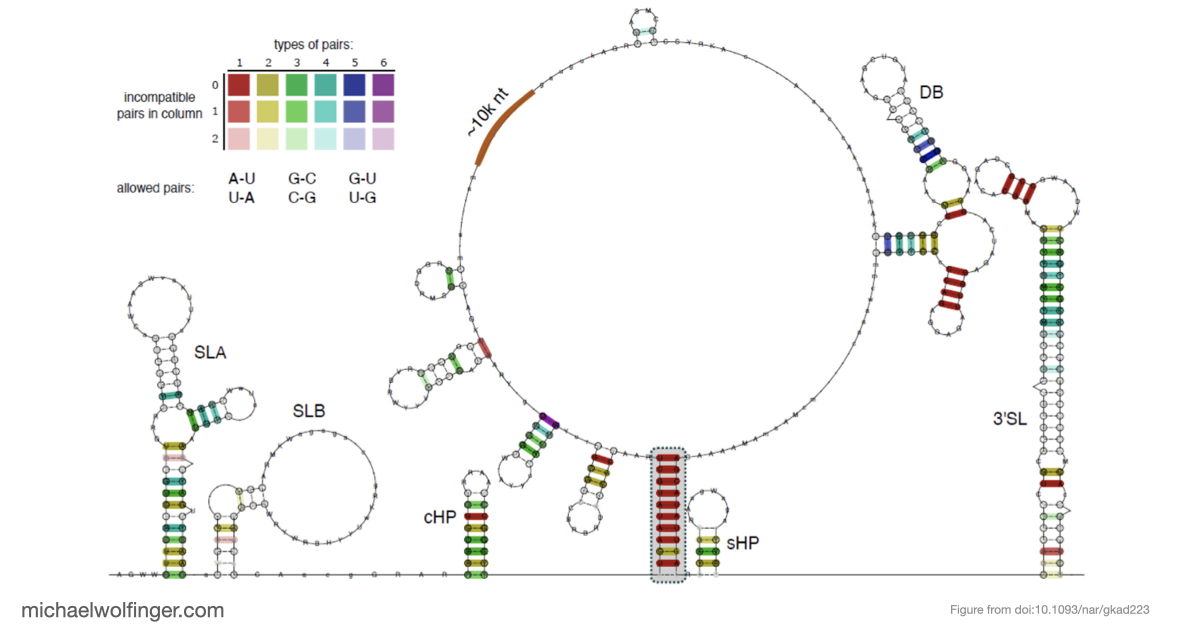
Long-range RNA-RNA interactions are central to the replication cycle of many flaviviruses, but they are not always easy to characterize directly. In Japanese encephalitis virus (JEV), the 5' and 3' terminal regions are expected to interact through conserved cyclization elements, yet the strength, stoichiometry, and structural behavior of that interaction still needed to be tested experimentally.
This study approaches the problem from both sides. Computational analysis was used to identify the most plausible interaction site and to evaluate whether the conserved cyclization sequence is kinetically favored over competing alternatives such as homodimer formation. The predicted interaction was then tested biophysically using SEC-MALS, analytical ultracentrifugation, microscale thermophoresis, and SAXS.
The result is a stronger case for the long-range interaction than computation alone could provide. The 5' and 3' terminal regions interact directly with nanomolar affinity and a 1:1 stoichiometry, and that interaction weakens substantially when the conserved cyclization sequence is disrupted. That is a useful mechanistic result because it ties the predicted long-range pairing to an experimentally measurable binding event.
Just as importantly, the kinetic analysis supports the idea that the cyclization sequence is not merely one possible pairing among many. It appears to be the dominant interaction pathway under the conditions examined, with an advantage over competing self-association states. For flavivirus biology, that matters because genome cyclization is closely tied to replication, and the computational ranking of alternative structures becomes much more informative once it is backed by binding measurements.
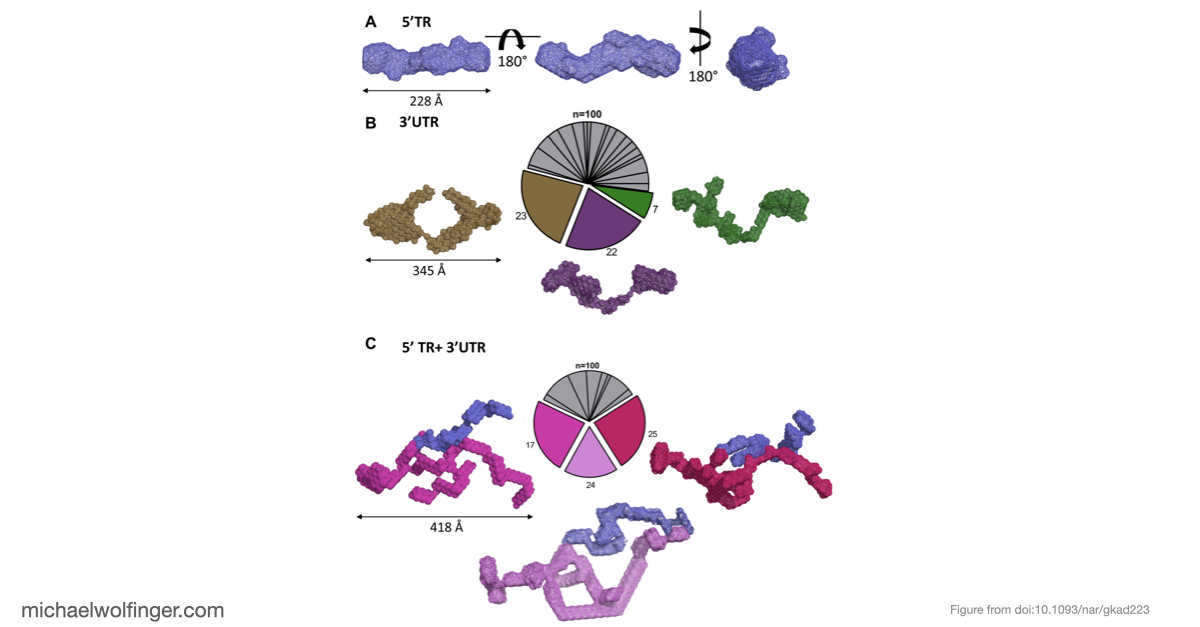
The SAXS-based structural model adds another piece of the picture. It does not deliver atomic resolution, but it does suggest that the complex is flexible while still maintaining a stable overall association in solution. That kind of low-resolution structural information is valuable when interpreting long viral RNAs, where conformational heterogeneity is often part of the biology rather than a nuisance to be averaged away.
More broadly, this paper is a good example of how RNA-RNA interaction studies should be done. Sequence analysis alone is too weak, and biophysical characterization without a clear computational hypothesis is inefficient. The combination is what makes the result persuasive: comparative and thermodynamic reasoning narrow the search space, and orthogonal biophysical methods test whether the predicted interaction actually exists and behaves as expected.
For teams working on viral RNA architecture, long-range RNA interactions, or structure-guided mechanistic questions, I also offer focused technical review and advisory support through my services page. Investigating RNA-RNA interactions through computational and biophysical analysisFigures and Data

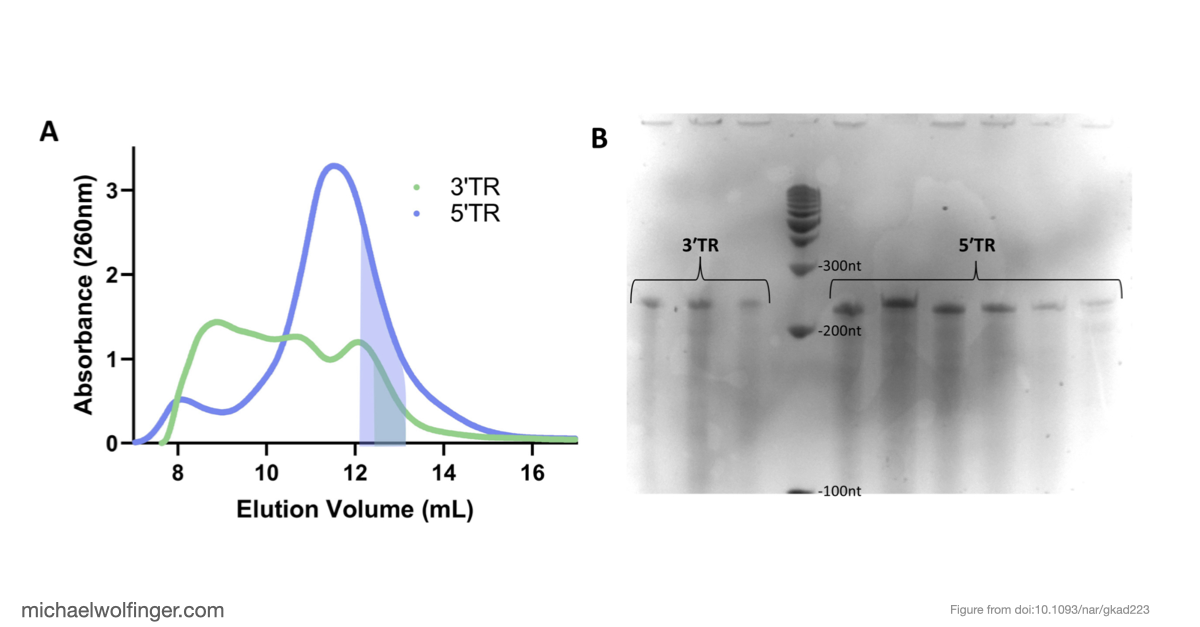
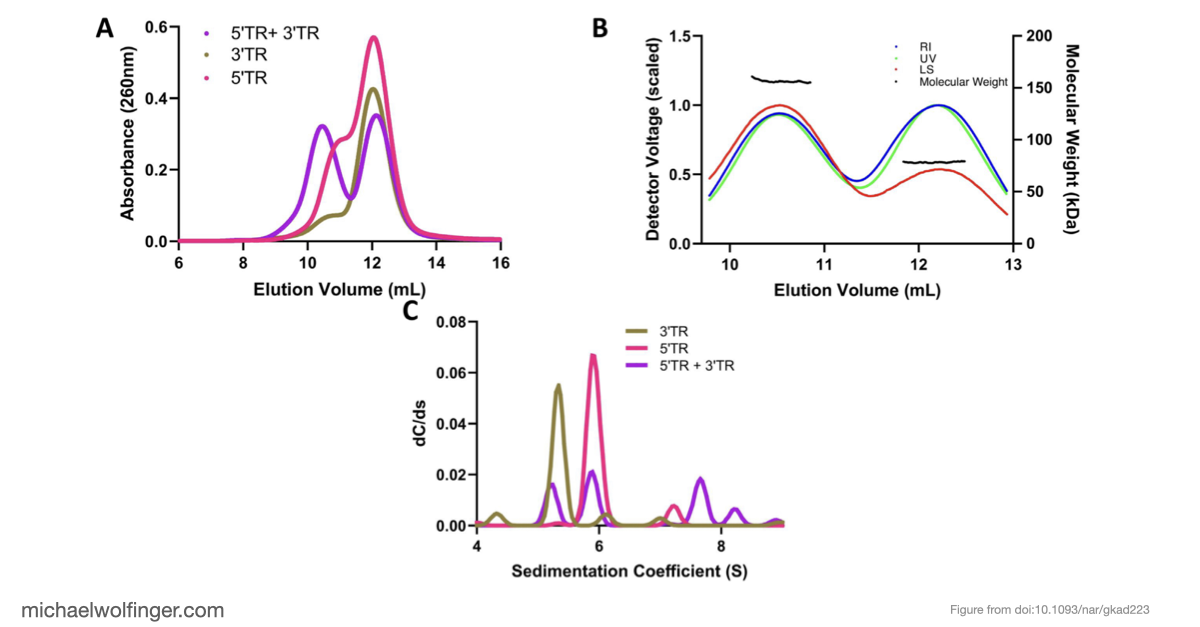
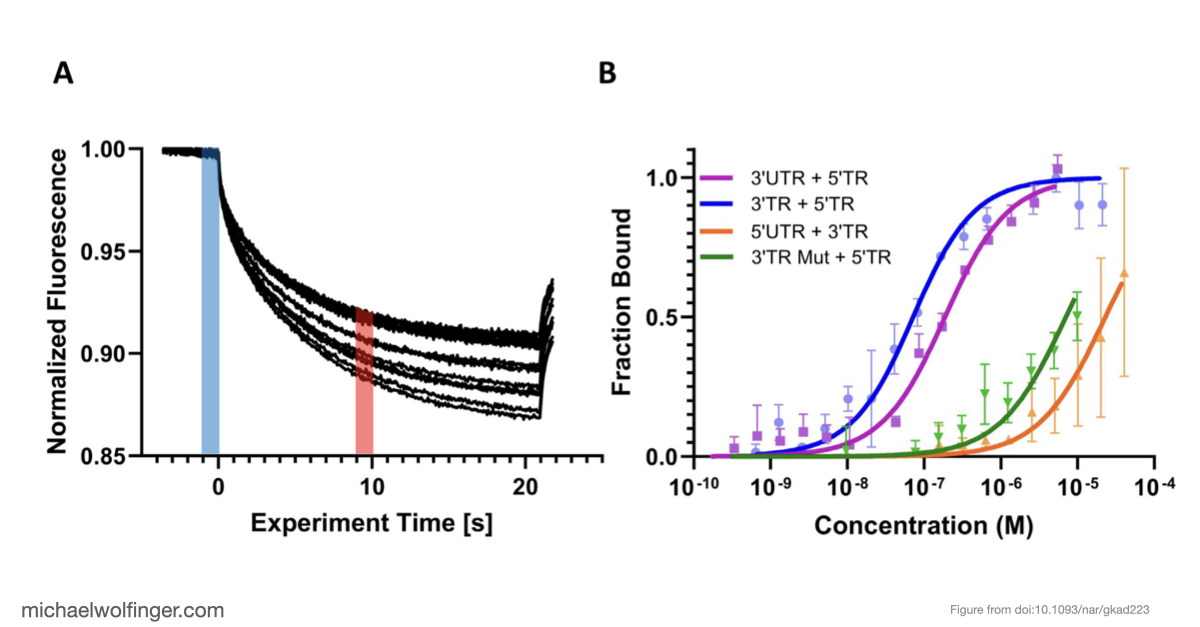
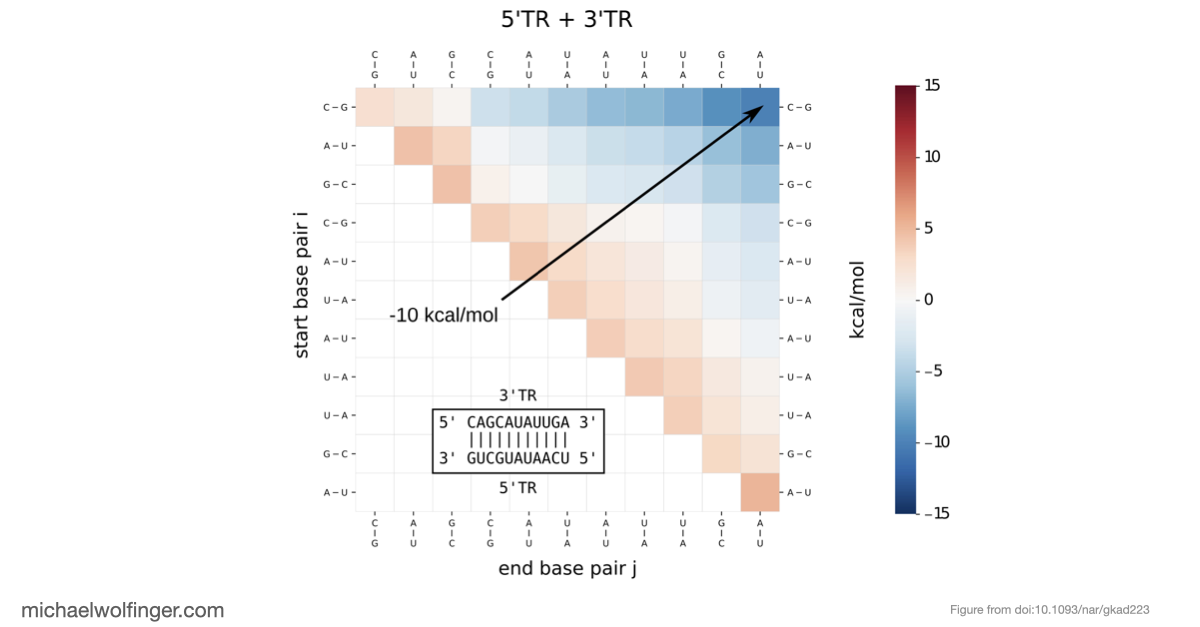
Citation
Tyler Mrozowich, Sean Park, Maria Waldl, Amy Henrickson, Scott Tersteeg, Corey R. Nelson, Anneke De Klerk, Borries Demeler, Ivo L. Hofacker, Michael T. Wolfinger, Trushar R. Patel
Nucleic Acids Res. gkad223 (2023) | doi:10.1093/nar/gkad223 | PDF | Supplement