RNA-protein complex refinement using AI modeling and docking
This study presents an efficient technique for refining protein-RNA complexes using artifilial intelligence (AI) based modeling and flexible docking. The method, utilizing parallel cascade selection molecular dynamics (PaCS-MD), accelerates conformational sampling of flexible RNA regions and produces high-quality complex models. Experimental validation demonstrates its superiority over template-based modeling, suggesting its potential for constructing complexes with non-canonical RNA-protein interactions
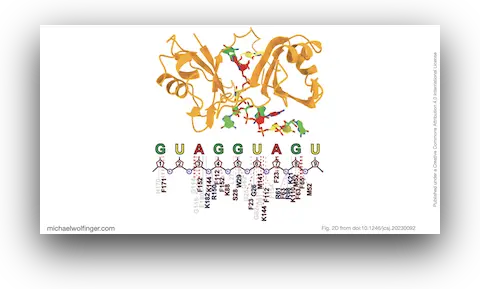
Understanding the interactions between proteins and RNA is essential for unraveling the mysteries of cellular functions. In a recent study, we proposed an innovative and efficient technique to accurately model the interactions between proteins and RNA, leading to insights that have the potential to advance the fields of structural biology and drug design.
When a protein interacts with RNA, it forms what is known as a protein-RNA complex. Constructing accurate models of these complexes is a formidable challenge. Traditional methods, such as template-based modeling, involve creating the overall conformation and then manually connecting RNA with a protein’s RNA-binding domains (RBDs). However, this doesn't always lead to biologically plausible complexes, highlighting the urgent need for improved methodologies to simulate protein-RNA association complexes.
As we were particularly interested in the RNA-binding protein Musashi-1 (MSI1) and its interaction with RNA through RNA-binding domains (MSI1-RBDs), we proposed a two-step approach: first, we utilized the AI-based modeling technique Alphafold2 to predict a 3D structure of the MSI1-RBDs based on the amino-acid sequences in humans. Following this, we used a flexible docking technique called PaCS-MD to construct the protein-RNA complexes. Unlike traditional methods that manually ligate RNA to the protein, PaCS-MD automatically constructs the complexes, providing a more natural interaction.
To ensure the validity of the constructed complexes, we evaluated their structural stability using the SIE (Solvation Interaction Energy) method. The results confirmed previous studies, showing that a core set of three nucleotides in MSI1-RBDs plays a significant role in the interaction with the target RNA. Moreover, we identified crucial residues for RNA-binding that aligned well with previous findings.
Our method offers a significant advantage over conventional techniques. Unlike traditional homology modeling methods, such as Phyre2, our technique captures the essential transitions required for forming protein-RNA complexes. However, we acknowledge that the technique relies on having initial structural information from NMR (Nuclear Magnetic Resonance) coordinates. Without this data, achieving accurate flexible docking can be challenging.
The study provides a compelling demonstration of constructing plausible protein-RNA complexes using a combination of AI-based modeling and flexible docking. This research takes us one step closer to deciphering the complex interactions between proteins and RNA, paving the way for advancements in personalized medicine and drug discovery.
Citation
A Structural Refinement Technique for Protein-RNA Complexes Using a Combination of AI-based Modeling and Flexible Docking: A Study of Musashi-1 Protein
Nitchakan Darai, Kowit Hengphasatporn, Peter Wolschann, Michael T. Wolfinger, Yasuteru Shigeta, Thanyada Rungrotmongkol, Ryuhei Harada
B. Chem. Soc. Jpn. (2023)
See Also
Theoretical studies on RNA recognition by Musashi 1 RNA–binding protein
Nitchakan Darai, Panupong Mahalapbutr, Peter Wolschann, Vannajan Sanghiran Lee, Michael T. Wolfinger, Thanyada Rungrotmongkol
Sci. Rep. 12:12137 (2022) | doi:10.1038/s41598-022-16252-w | PDF | Figures
Musashi Binding Elements in Zika and Related Flavivirus 3’UTRs: A Comparative Study in Silico
Adriano de Bernardi Schneider, Michael T. Wolfinger
Sci. Rep. 9(1):6911 (2019) | doi:10.1038/s41598-019-43390-5 | PDF | Figures